MACSE
This is the documentation for MACSE v2.00
1. Overview
MACSE (Multiple Alignment of Coding SEquences Accounting for Frameshifts and Stop Codons) provides a complete toolkit dedicated to the multiple alignment of coding sequences that can be leveraged via both the command line and a Graphical User Interface (GUI).
Multiple sequence alignment (MSA) is a crucial step in many evolutionary analyses. Nonetheless, most existing alignment tools ignore the underlying codon structure of protein-coding nucleotide sequences. Accounting for this structure is not only useful to improve the proposed alignment, but it is also a prerequisite for some downstream analyses such as detection of selection footprints based on the ratio of non-synonymous to synonymous substitutions (dN/dS).
MACSE aligns protein-coding nucleotide (NT) sequences with respect to their amino acid (AA) translation while allowing NT sequences to contain multiple frameshifts and/or stop codons. MACSE was hence the first automatic solution to align protein-coding gene datasets containing non-functional sequences (pseudogenes) without disrupting the underlying codon structure. It has also proved useful in detecting undocumented frameshifts in public database sequences and in aligning next-generation sequencing reads/contigs against a reference coding sequence.
In the output alignment produced by MACSE, frameshifts are indicated using ‘!’. You can specify to MACSE a subset of your sequences that are more likely to contain frameshifts or stop codons, the less reliable sequences in the MACSE terminology. This allows to use a lower cost when introducing a stop codon or a frameshift in those sequences as compared to introducing such events in other reliable sequences. More details and examples concerning this feature are provided in the alignSequences section.
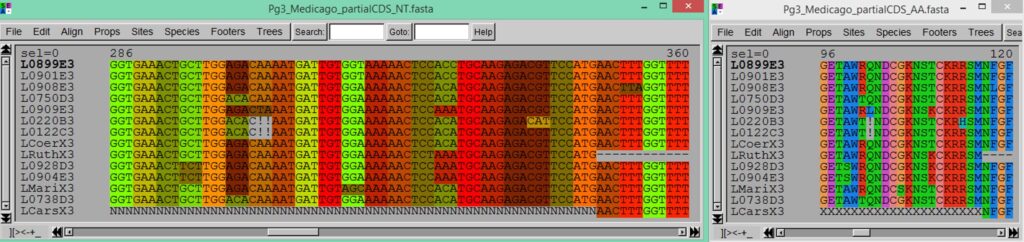
Seaview is very convenient to visualize sequence alignments produced by MACSE. Indeed, Seaview accepts the ‘!’ character in both nucleotide and amino acid sequences and also allows to visually emphasize the codon structure of the aligned nucleotide sequences.
2. Getting started with MACSE
If you are new to MACSE, you should probably start here:
3. Programs
- alignSequences: aligns nucleotide (NT) coding sequences using their amino acid (AA) translations.
- alignTwoProfiles: aligns two previously computed alignments (also called profiles).
- enrichAlignment: adds sequences to a previously computed alignment.
- exportAlignment: exports alignments (different formats) and other output files.
- multiPrograms: sequentially executes multiple MACSE commands contained in a text file (one per line).
- refineAlignment: improves a previously computed alignment.
- reportGapsAA2NT: reports gaps from aligned AA sequences to the corresponding (unaligned) NT sequences.
- reportMaskAA2NT: uses a NT alignment and a filtered (masked) version of its AA translation to derive the NT alignment.
- splitAlignment: splits an alignment to extract a subset of selected sequences and/or sites.
- translateNT2AA: translates protein-coding nucleotide sequences into amino acid sequences.
- trimAlignment: trims the input alignment by removing gappy sites at the beginning/end of the alignment.
- trimNonHomologousFragments: identifies sequence fragments that do not share homology with other sequences and remove those fragments.