MACSE
Deriving a nucleotide alignment from an amino acid alignment
The alignSequences subprogram could be time consuming for large datasets. A possible, still efficient, strategy for large datasets is the following:
- 1. use trimNonHomologousFragments to eliminate non-homologous sequence fragments
- 2. use alignSequences with rapid optimization options to unravel frameshifts
- 3. preserve frameshifts (but not gaps) to obtained unaligned nucleotide sequences with documented frameshifts (simply delete the ‘-‘ from your FASTA file)
- 4. translate those nucleotide sequences into amino acid sequences using translateNT2AA.
- 5. align these amino acids sequences with your favorite alignment software (e.g. MUSCLE, PRANK, MAFFT)
- 6. use reportGapsAA2NT to derive your nucleotide alignment from the amino acid alignment found at step 5.
This pipeline as been successfully used to produce OrthoMaM alignments and is available through a dedicated web service at http://mbb.univ-montp2.fr/MBB/.
1. Reporting gaps from aligned amino acid sequences to unaligned nucleotide sequences
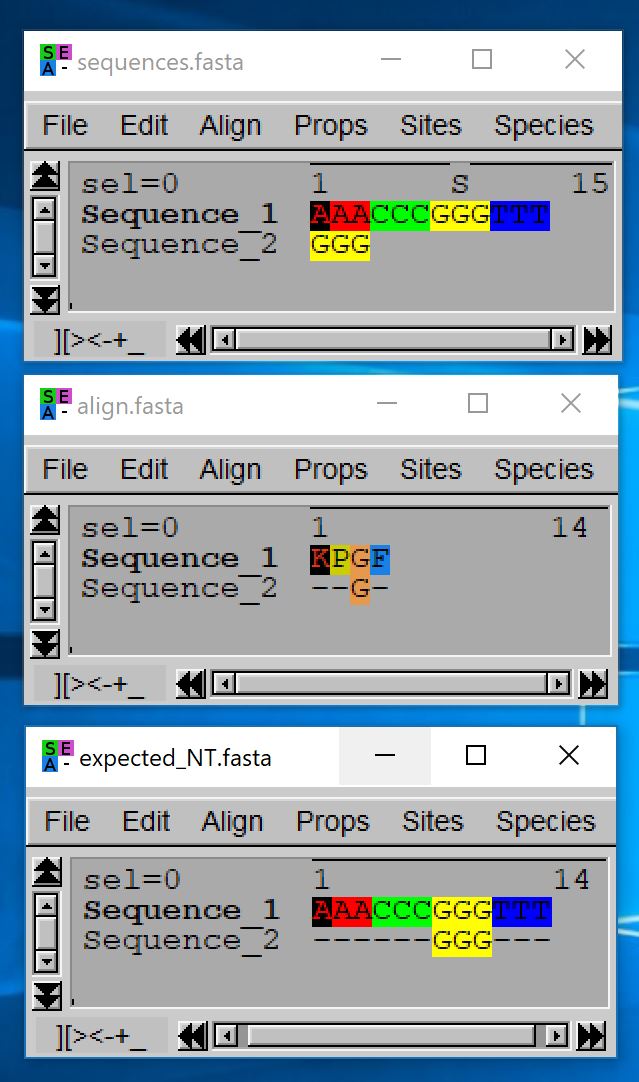
The reportGapsAA2NT takes as input a FASTA file with unaligned nucleotide sequences and a FASTA file of aligned sequences that are the amino acid translations of the nucleotide ones. Each sequence should hence be present with the exact same name in both files and should be three time longer in the nucleotide file than in the amino acid file (ignoring gaps).
Warning: For the sequence lengths to match, you should either remove any final stop codons from your nucleotide sequences or translate them into the unkwown amino acid ‘X’.
URL : samples/reportGapsAA2NT/
- java -jar macse.jar -prog reportGapsAA2NT -align_AA align.fasta -seq sequences.fasta
- java -jar macse.jar -prog reportGapsAA2NT -align_AA align.fasta -seq sequences.fasta -out_NT output_NT.fasta
2. Reporting gaps from a sequence pattern to a nucleotide alignment
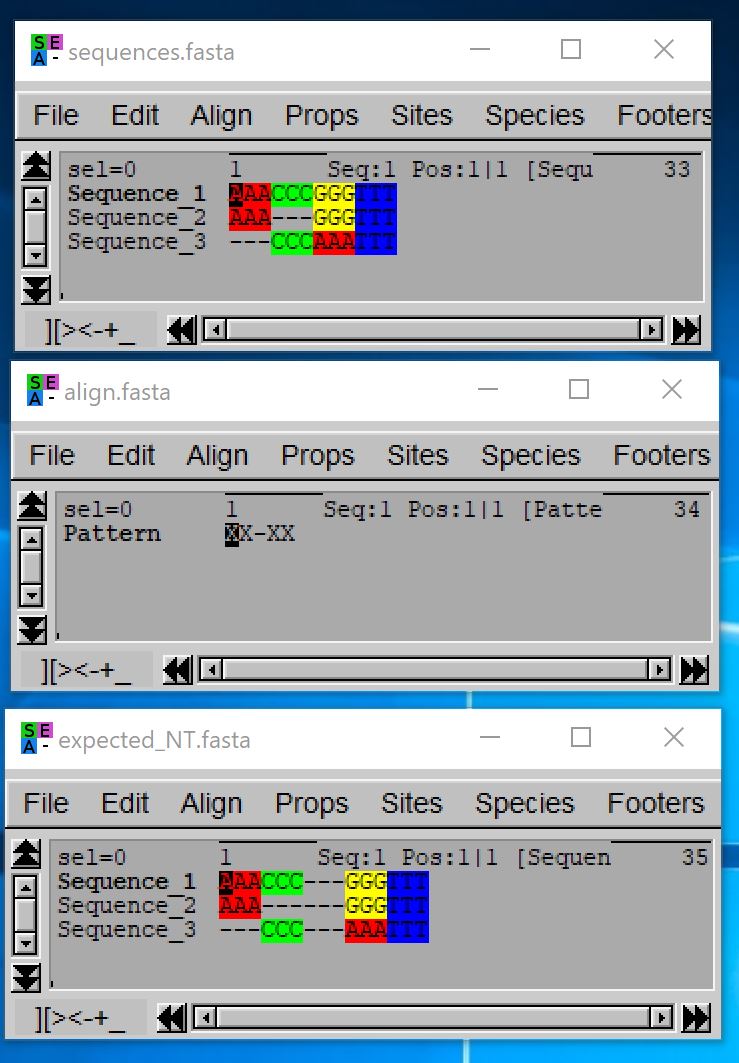
This option could be useful to build an alignment with a very large number of sequences: you can start by clustering them (either based on homology or taxonomy), then aligning independently each cluster of sequences and produce a codon consensus sequence for each aligned cluster (exportAlignment, align those consensus sequences, and use the resulting alignment to produce the global alignment by reporting gaps of each consensus sequence into the corresponding cluster alignment. In such case, the aligned consensus sequence is thus seen as a pattern indicating where gaps should be added to the cluster alignment in order to build the global alignment.
- java -jar macse.jar -prog reportGapsAA2NT -align_AA align.fasta -seq sequences.fasta -AA_seq_as_pattern_ON
Warning: with this option, the sequences.fasta file must contain aligned nucleotide sequences and the align.fasta file must only contain one sequence that serves as pattern to indicate where gaps should be inserted in the input nucleotide alignment:
3. Related documentation
You can find other options related to this program from the following link: