MACSE
Sequence alignment
The MACSE subprogram alignSequences aligns protein-coding sequences at the nucleotide level while scoring the considered nucleotide alignments based on their amino acid translation. It thus favours nucleotide gap stretches that are multiple of three but also considers those inducing frameshifts, when they allow to recover the underlying codon structure. MACSE therefore produces alignments which benefit from the higher similarity of amino acid sequences while accounting for frameshifts and stop codons that could occur in pseudogenes or in poor quality sequences (alignment costs).
1. Basic usage
Folder: samples/alignSequences/
The alignSequences program aligns protein-coding nucleotide sequences provided in a FASTA file. Here is a basic example of its usage on a subset of the TMEM184 CDS provided in the release 59 of the EnsEMBL database:
java -jar macse.jar -prog alignSequences -seq TMEM184_Ensembl_small.fastaIn this example, MACSE aligns nucleotide sequences found in the TMEM184_Ensembl_small.fasta file using MACSE default parameters. The resulting nucleotide alignment is saved in TMEM184_Ensembl_small_NT.fasta and the corresponding amino acid alignment is saved in TMEM184_Ensembl_small_AA.fasta. Note that, in this example, using MACSE reveals undocumented frameshifts in Pan and Pongo sequences (traceable by the character ‘!’). Those ‘frameshifts’ are probably due to annotation errors, more details in Ranwez et al 2011, Fig 4.
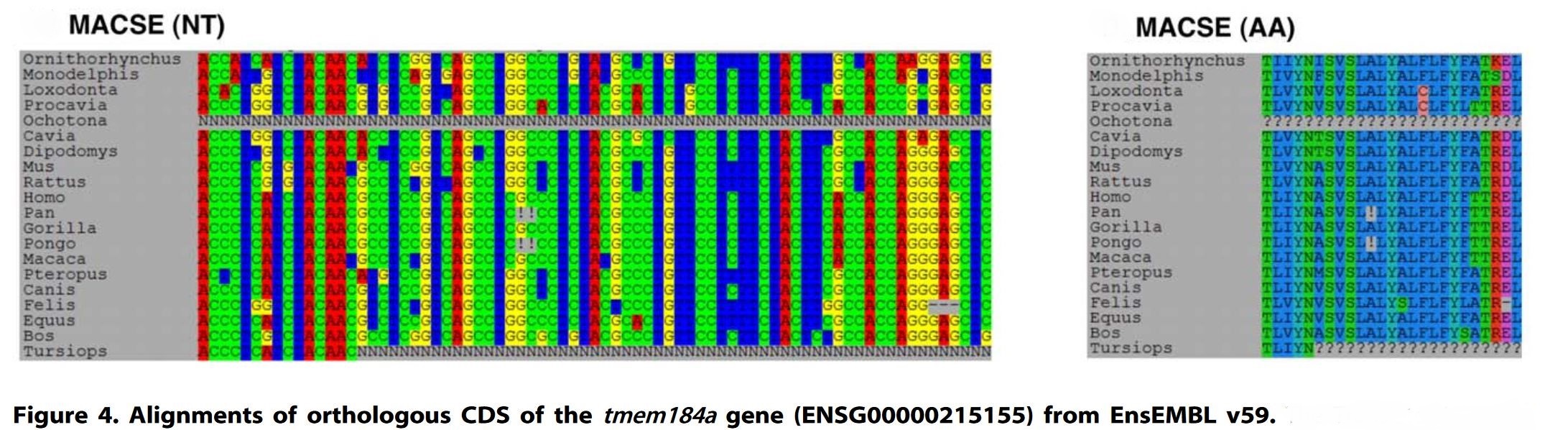
The seq option is the only mandatory option for this MACSE subprogram. Any gaps present in the input FASTA file will be ignored, if you want to improve an existing alignment you should instead use the refineAlignment subprogram.
2. Changing default output file names
Folder: samples/alignSequences/outputs/
The names of the fasta alignment files created by MACSE can be specified using out_NT and out_AA options as follow:
java -jar macse.jar -prog alignSequences -seq sequences.fasta -out_NT output_NT.fasta -out_AA output_AA.fasta3. Less reliable sequences
Folder: samples/alignSequences/seq_lr/
In case you think some sequences are more likely to contain frameshifts and/or stop codons than others, it is probably better to use different frameshift and stop codon costs for those less reliable (lr) sequences. MACSE allows you to do treat those less reliable sequences differently by simply putting them in a different fasta file, and using the seq_lr option to indicate the name of the fasta file containing them:
- java -jar macse.jar -prog alignSequences -seq AMBN_coding.fasta -seq_lr AMBN_pseudo.fasta
In this example, we suspect that some AMBN sequences involved in tooth enamel structure are pseudogenes in toothless species. We thus split our sequences in two files AMBN_coding.fasta and AMBN_pseudo_lr.fasta, so that MACSE could align all those sequences together using a smaller penalty for inserting frameshifts and stop codons in the pseudogene sequences.
Differentiating reliable from less reliable sequences is especially relevant when your dataset contains, for instance, reference sequences from model organisms found in public database and newly annotated sequences or pseudogenes.
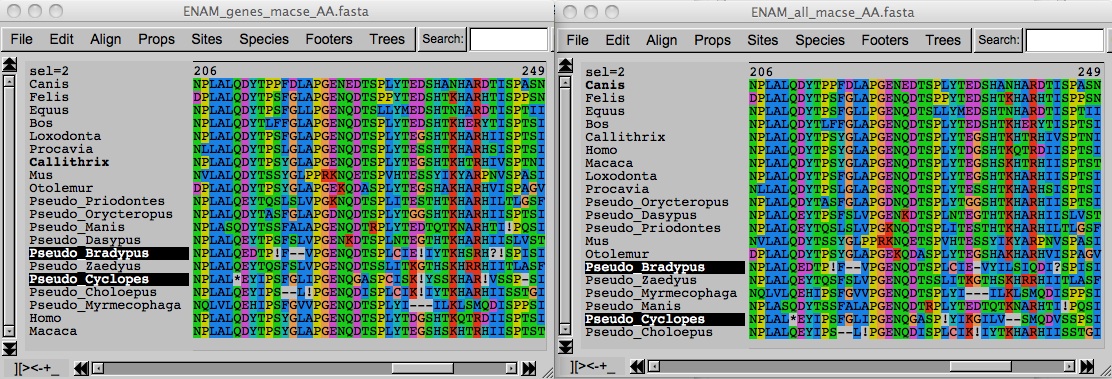
If you just suspect pseudogenization, you can use a two step approach. In a first step, you can run alignSequences with a single fasta file (and faster optimization, see below) and use the first draft alignment obtained to identify possible pseudogene sequences. Then, in a second step, you can re-launch alignSequences using the option seq_lr with a fasta file containing those identified putative pseudogenes.
4. Controlling degree of optimization and speed
Folder: samples/alignSequences/maxRefines/
You should be aware that MACSE could be time consuming. If you just want a rapid first draft of your alignment (e.g. to check if your sequences potentially have frameshifts) or if you know that your sequences are easy to align since highly similar, you can speed up the alignment process.
Once having a first multiple sequence alignment (MSA), MACSE uses a classical 2-cut refinement strategy to improve it. This strategy consists in partitioning the current alignment into two sub-alignments that are subsequently re-aligned. The resulting MSA replaces the previous one if its score is improved. This 2-cut refinement strategy also uses the guide tree: it iteratively considers each clade of the guide tree and splits the current global alignment so that one of the two sub-alignments contains the exact sequences of the concerned clade. Once all (external) branches have been tested, if the score has been improved, the guide tree is updated and a new iteration of this refinement loop will start.
Note that once you have an alignment, if you split it in two sub-alignments you can keep track of the relative position of each sites of the first alignment compared to the second one. By default MACSE uses this information to go a little bit faster by limiting its search.
Four options allow to control the balance between optimization speed and alignment accuracy:
– optim allows you to control whether you use standart 2-cut (default), only remove and realign one sequence at a time (-optim 1) or only want to know the score of your alignment (-optim 0 no refinement at all).
– max_refine_iter allows you to define the maximum number of refinement iterations, i.e. how many time the split/re-alignments induced by the guide tree (external) branches will be tested in the worst case.
– local_realign_init if smaller than 1 the first refinement loop will consider only local improvement (the lower this value the faster the initial refinement, possible values [0-1]).
– local_realign_dec if smaller than 1 each refinement loop will be faster than the previous one by focusing on a smaller interval during alignment improvement steps (the lower this value the faster the refinements, possible values [0-1])
For instance to get the first draft alignment done by MACSE, prior to any optimization, use this command line:
- java -jar macse.jar -prog alignSequences -seq TMEM184_Ensembl_Plos.fasta -max_refine_iter 0
If you want a relatively quick optimization, you can try for instance:
- java -jar macse.jar -prog alignSequences -seq TMEM184_Ensembl_Plos.fasta -max_refine_iter 3 -local_realign_init 0.3 -local_realign_dec 0.2
If you want an extensive search equivalent to what was done in MACSE v1 use:
- java -jar macse.jar -prog alignSequences -seq TMEM184_Ensembl_small.fasta -local_realign_init 1 -local_realign_dec 1
5. Other options (genetic codes, alignment costs, AA alphabets, etc…)
You can find other options related to this program from the following links: