MACSE
Alignment cost parameters
MACSE defines the score/cost of a nucleotide alignment based on its amino acid translation. The cost of an alignment depends on the elementary costs of the events it encompassed:
- the bonus/cost of one amino acid facing another is defined in the amino acid substitution matrix (default or specified, cf 7:alternative substitution matrix)
- the cost of a stretch of n consecutive gaps is defined as 1/ a fix cost for opening this gap stretch plus 2/ an extension cost proportional to its length n
- the cost of a frameshift and a stop codon may vary depending on 1/ whether they are inside the sequence or at its extremities and 2/ the type of sequence where they appear (reliable or not, see seq_lr option example)
MACSE provides some default values, but you can set all those costs at another value. Note that:
- if you enter a positive value for a cost it will automatically be converted into negative values (you do not need to worry about signs).
- you can use up to one decimal for costs.
1. names of the cost options
- cost options related to gap, frameshift and stop codon have names starting by gap, FS and stop respectively.
- gap cost options related to opening or extension have op or ext in their names
- cost options specific to less reliable sequences have lr in their names
- penalties for events appearing at the end(s) of a sequence have term in their names
- term stands for terminal and refers to the end of a sequence, indifferently to the beginning or the end for gaps and frameshifts, but only to the end of the sequence for stop codons.
2. specifying gap costs and fixing unexpected results
- gaps are equally penalized in all sequences, reliable (-seq) or not (-seq_lr).
- usually gaps at the beginning or at the end of the alignment are less penalized as they can simply result from the fact that the locus is not completely sequenced (different primer set or shorter contig).
In MACSE options, gaps located between the first and the last amino acid are called internal gaps whereas those before the first amino acids or after the last one are called terminal gaps.
Folder: samples/delegations/costs/
You can adjust all or some gap options using the following options:
for gap opening within the sequence:
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -gap_op 15
for terminal gaps opening (gaps not within the sequence):
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -gap_op_term 20
for gaps extension within a sequence
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -gap_ext 10
for terminal gaps extension (gaps not within the sequence):
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -gap_ext_term 10
Some guidelines for gaps:
- If you have too many small gaps, you can try to increase the penalty of gap opening (this would favor alignment with fewer but longer gaps).
- If you have some incomplete sequences you can try to decrease the penalty of external gaps.
- There is unfortunately no perfect way to fix those costs and this is one of the key challenges of multiple sequence alignment software.
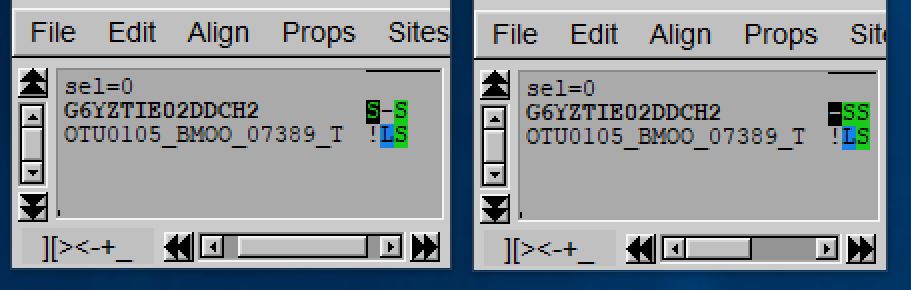
Even with a very simple dataset, you can have results that seems unexpected. By default MACSE penalizes only slightly more internal gaps than terminal ones. This could produce that seems unexpected as the one below (Folder:samples/delegations/costs/unexpected/):
- java -jar macse.jar -prog alignSequences -seq small_weird.fasta
A very different result could be obtained by penalizing less external gaps:
- java -jar macse.jar -prog alignSequences -seq small_weird.fasta -gap_op 7 -gap_op_term 3.5
The figure below displays the alignment obtained with default costs (on the left) and the one obtained when penalizing less external gap opening (on the right). The alignment on the right has a penalty of -4 due to the L (leucine) facing the S (Serine) that the one of the right do not have by default the bonus of having an internal gap instead of an external gap is not enough to compensate this -4 penalty, and the alignment on the left is favor (with a slight score difference -14 vs -14.3).
3. Specifying frameshift cost
Frameshift can be seen as an unexpected/unlikely kind of gap stretch (a nucleotide gap stretch that is not a multiple of 3 in a coding nucleotide sequence). They could be real biological events (e.g. in pseudogenes, or in sequence that will be post-processed by the molecular machinery) or due to sequencing/annotation errors. While MACSE allows such events, they are strongly penalized in reliable sequences, as they are unexpected in nucleotide coding sequences, and a little less in less reliable sequences.
By default, a frameshift in the first or last codon is not penalized by MACSE as this usually reflects an incomplete sequence or a UTR annotation error that are quite frequent.
Folder: samples/delegations/costs/fs/
You can specify 4 different frameshift costs for internal vs external frameshifts, appearing in reliable vs less reliable sequences:
Internal frameshifts in reliable sequences
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -fs 5
Terminal frameshifts in reliable sequences
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -fs_term 30
Internal frameshifts in less reliable sequences
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -seq_lr sequences_lr.fasta -fs_lr 15
Terminal frameshifts in less reliable sequences
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -seq_lr sequences_lr.fasta -fs_lr_term 20
4. Internal stops
By default stop codons are not penalized at the end of a sequence; they are penalized everywhere else.
As stop codons are expected at the end of the sequences their cost is always null. You can only specify 2 different stop costs for internal stops appearing in reliable vs less reliable sequences:
specifying cost for stop condons within reliable sequences
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -stop 10
specifying cost for stop condons within less reliable sequences
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -seq_lr sequences_lr.fasta -stop_lr 40
Warning concerning FS and stop costs:
For a given sequence type a stop codon should never cost more than two frameshifts, otherwise all stop codons will be avoided by MACSE by including two successive frameshifts.
5. Aligning functional and non-functional sequences (pseudogenes).
If you know, a priori, which sequences are functional protein-coding sequences and which ones are non-functional (i.e. pseudogenes), it is preferable to have them in different files (as described in ‘Sequence alignment 3. Less reliable sequences’). You can then ask MACSE to use different costs for each file, assigning standard costs to the file containg functional protein-coding sequences (option fs and stop) and lower costs for the other file, containing the less reliable sequences (option fs_lr and stop_lr).
In pseudogenes a STOP can result from a single mutation whereas frameshifts require one insertion/deletion. It thus seems reasonable to not penalize stop codons more than frameshifts in such sequences, e.g. using a penalty between 10 and 30 for both events.
For pseudo-genes -fs_lr 10 -stop_lr 10 is often a good initial parameter set.
- java -jar macse.jar -prog alignSequences -seq ENAM_genes.fasta -seq_lr ENAM_pseudos.fasta -fs_lr 10 -stop_lr 10
Note that you can test other values (e.g. between 10 and 30) since the frequency of frameshifts ans stops in your pseudogene sequences (hence the relevant cost) is related to the time elapsed since your sequences are no longer coding.
- java -jar macse.jar -prog alignSequences -seq Song_PNAS2008_Grasshoppers_COX1_genes.fasta -seq_lr Song_PNAS2008_Grasshoppers_COX1_pseudo.fasta -gc_def 5 -stop_lr 30 -fs_lr 30
6. Aligning NGS data (reads or contigs).
When aligning a set of sequences containing NGS data it is preferable, as for pseudogenes, to split the sequences in two files and to use different costs for frameshifts and stop codons in order to account for potential sequencing errors in NGS reads or contigs. For NGS raw sequences (reads or contigs) -fs_lr 10 -stop_lr 15 is often a good initial parameter set. You can of course adapt these costs depending on the sequencing technology used since some technologies induce more frequent errors in homopolymers (e.g. 454) seen as frameshifts by MACSE, while others rather induce nucleotide calling errors leading to potential stop codons. The ideal case is to have a set or reliable sequences (i.e. from public databases or manually annotated) that you can provide as reference to MACSE in order to guide the alignment process. In this exemple, we identified, by BLAST, reads similar to Ensembl sequences ENSG00000119777 (TMEM214) and used orthologous CDS sequences to guide the alignments of 454 contigs while accounting for probable sequencing errors:
- java -jar macse.jar -prog alignSequences -seq TMEM214.fasta -seq_lr TMEM214_reads.fasta -fs_lr 10 -stop_lr 15
Note that the more reliable sequences the better. If you include numerous reliable sequences or have a clean alignment corresponding to your current contigs, you can use the enrichAlignment to rapidly add new contigs to your initial alignment (see corresponding sections for detailed examples).
7. Using an alternative substitution matrix
Folder: samples/delegations/costs/scoreMatrix/
- java -jar macse.jar -prog alignSequences -seq sequences.fasta -score_matrix VTML240
Available substitution matrix are:
- BLOSUM62
- VTML200BIS
- VTML240
Note that all default costs are set for the Blosum62 matrix. If you decide to use a different matrix you should probably use different costs for all other options.