MACSE
Refining alignments
The refineAlignment subprogram tries to further improve an existing nucleotide alignment. It aligns sequences at the nucleotide level while scoring the considered nucleotide alignments based on their amino acid translation. It thus favors nucleotide gap stretches that are multiple of three but it also considers those inducing frameshifts, when they allow to recover the underlying codon structure. It therefore produces alignments which benefit from the higher similarity of amino acid sequences while accounting for frameshifts and stop codons that could occur in pseudogenes or in poor quality sequences (alignment costs).
This program is highly related to alignSequences. The main difference is that alignSequences takes unaligned sequences as input and thus need to build a first alignment before improving it, while refineAlignment starts from an alignment as input. Those two programs share most of their options and it is strongly advised to read the alignSequences help pages before using refineAlignment.
refineAlignment can be used to improve an alignment produced by MACSE (e.g. produced by alignSequences with prameters favoring speed over accuracy) or by any other alignment software.
- On one hand, if you used a 3-step approach to align your sequences (1/ translating all coding NT into AA, 2/ aligning those AA sequences, and 3/ using the obtained amino acid alignment to derive the NT one) then efineAlignment can be relevant to rapidly identify sequences with frameshits that have been erroneously aligned using this strategy. We however strongly suggest to use alignSequences with adequate speed options to spot frameshifts prior to using this 3 steps strategy.
- On the other hand, if you have simply aligned your sequences at the nucleotide level, the resulting alignment can be so bad (at least in regard to the underlying codon structure) that refineAlignment will be very slow and may even worsen the input alignment.
Folder: samples/refineAlignment/
1. Some basic usage examples
The same options as those used for alignSequences allow you to (1) control for the balance between optimization and speed (-optim, -max_refine_iter, local_realign_init, local_realign_dec), (2) specify a subset of less reliable sequences with different costs for their frameshifts and stop codons (-seq_lr), (3) choose the name of output files (out_NT, out_AA), and (4) select your own elementary alignment costs:
- java -jar macse.jar -prog refineAlignment -align align.fasta -optim 1
- java -jar macse.jar -prog refineAlignment -align align.fasta -optim 2
- java -jar macse.jar -prog refineAlignment -optim 2 -align align.fasta -out_NT output_NT.fasta -out_AA output_AA.fasta
- java -jar macse.jar -prog refineAlignment -align align.fasta -optim 2 -seq sequences.fasta -seq_lr sequences_lr.fasta
2. A more realistic example related to metabarcoding
Metabarcoding analysis often requires to handle thousands of sequences. Such datasets are not directly tractable with the alignSequence subprogram of MACSE, but they can be handled by sequentially adding your newly obtained sequences to a reference alignment containing protein-coding sequences of related taxa for your targeted locus (COX1, matK, rbcL, etc…). We successfully used this approach in the Moorea project, Leray et al. 2013. In this project, we initially got an external alignment (from previous studies) of about 7,000 COX1 sequences. In a fisrt step, we used refineAlignment to identify potential errors in this alignment. About 200 sequences were detected as having unexpected frameshifts or stop codons. In the following example, the analysis is shown on a small subset of the original alignment:
- java -jar macse.jar -prog refineAlignment -align Moorea_BIOCODE_small.fasta -gc_def 5 -optim 1 -max_refine_iter 1
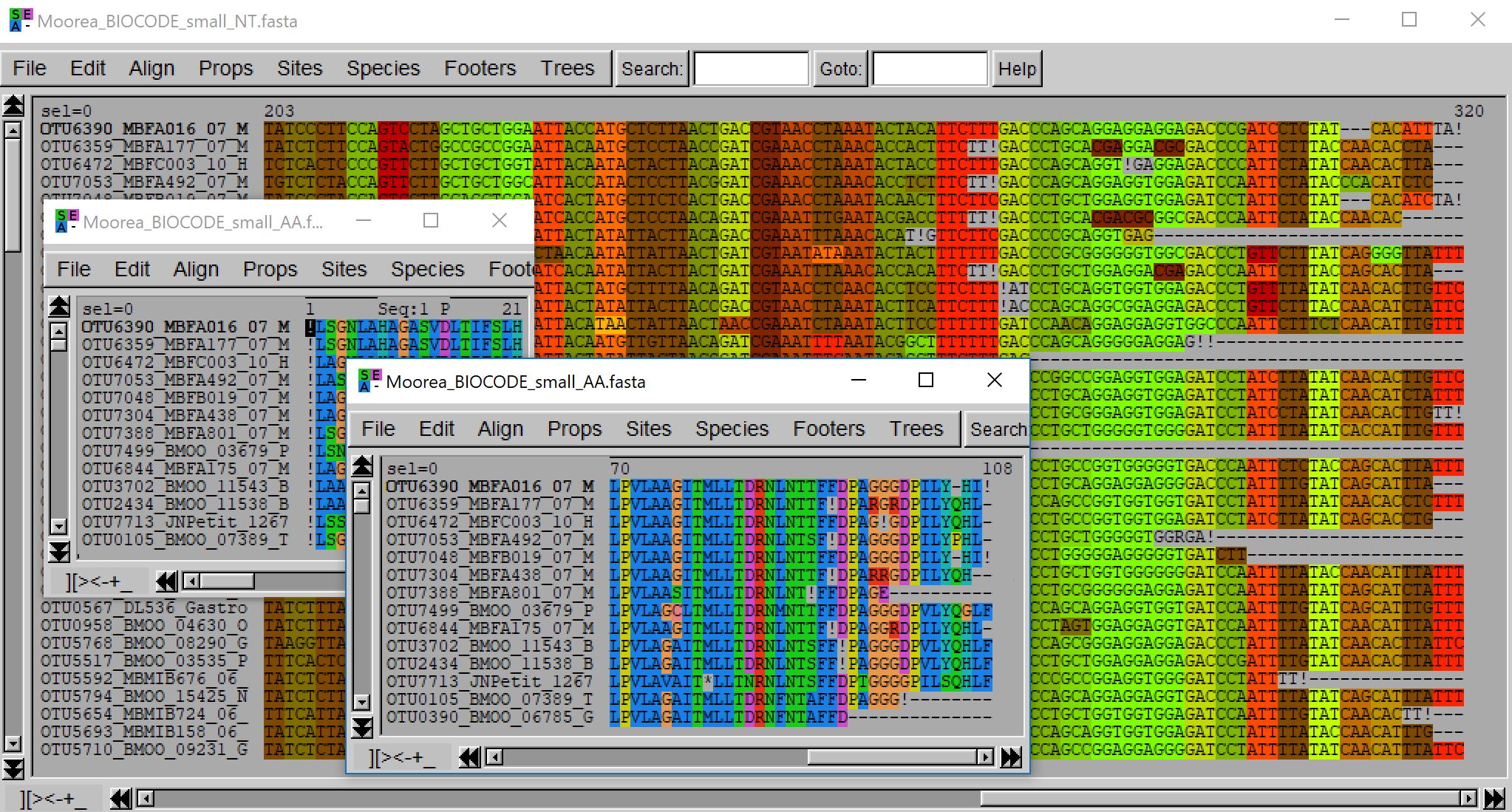
In this example, the input alignment mostly respects the codon structure but it started on the third reading frame. refineAlignment automatically detects this and adds the required frameshits at the beginning of the sequences. Moreover, it improves the alignment of a dozen of sequences (appearing together in the fasta file and hence easier to spot) by introducing some frameshifts near the sequence ends. Those sequences, with few frameshifts and stop codons, can then be kept or (manually) removed depending on your objective.
The frameshifts at the extremities of the sequences can be replaced by gap codons using exportAlignment. This is probably better if all first or all last codons are frameshifts since such configurations can distorted the count of frameshift events done by enrichAlignment when conditionally adding new sequences.
- java -jar macse.jar -prog exportAlignment -align Moorea_BIOCODE_small_ref.fasta -codonForExternalFS — -codonForInternalFS —
3. Related documentation
You can find other options related to this program from the following links: