MACSE
Alignment export
MACSE pinpoints frameshifts using the « ! » character. However this is not standard usage and alignment with such characters will be rejected by most software that take a multiple sequence alignment as input. This MACSE subprogram allows to replace « ! » characters in nucleotide and amino acid alignments. It also allows computing some basic statistics about the number insertions, deletions, frameshifts, and stop codons per sequence.
This export procedure as several advantages over a simple « search and replace » of the « ! » character in your FASTA files:
– 1. you can choose to modify the whole codons containing frameshifts (e.g. replacing AT! by either — or NNN) rather than just acting on a single character (e.g. replacing AT! by either AT- or ATN) – 2. you can handle differently frameshifts that occur within sequences and those at sequence extremities – 3. you can choose to transform internal stop codons into NNN codons – 4. if after replacement you have useless sites entirely made of gap-only codons (—) those sites will be removed.
URL : samples/exportAlignment/
1. Replacing frameshifts and stop codons
As a stop codon is expected at the end of full coding sequences, you can handle differently the stop codons appearing at the end of the sequences and those appearing within the sequences.
- java -jar macse.jar -prog exportAlignment -align align.fasta -codonForFinalStop —
- java -jar macse.jar -prog exportAlignment -align align.fasta -codonForInternalStop NNN
Similarly, terminal frameshifts (those at the end of the sequences) often reflect the fact that the coding sequence is incomplete rather than the fact that it contains an error or a frameshift. You can hence handle them differently.
- java -jar macse.jar -prog exportAlignment -align align.fasta -codonForExternalFS —
- java -jar macse.jar -prog exportAlignment -align align.fasta -codonForInternalFS NNN
Finally, you can specify how to handle frameshift characters that still remain after replacement performed at the codon level.
- java -jar macse.jar -prog exportAlignment -align align.fasta -charForRemainingFS –
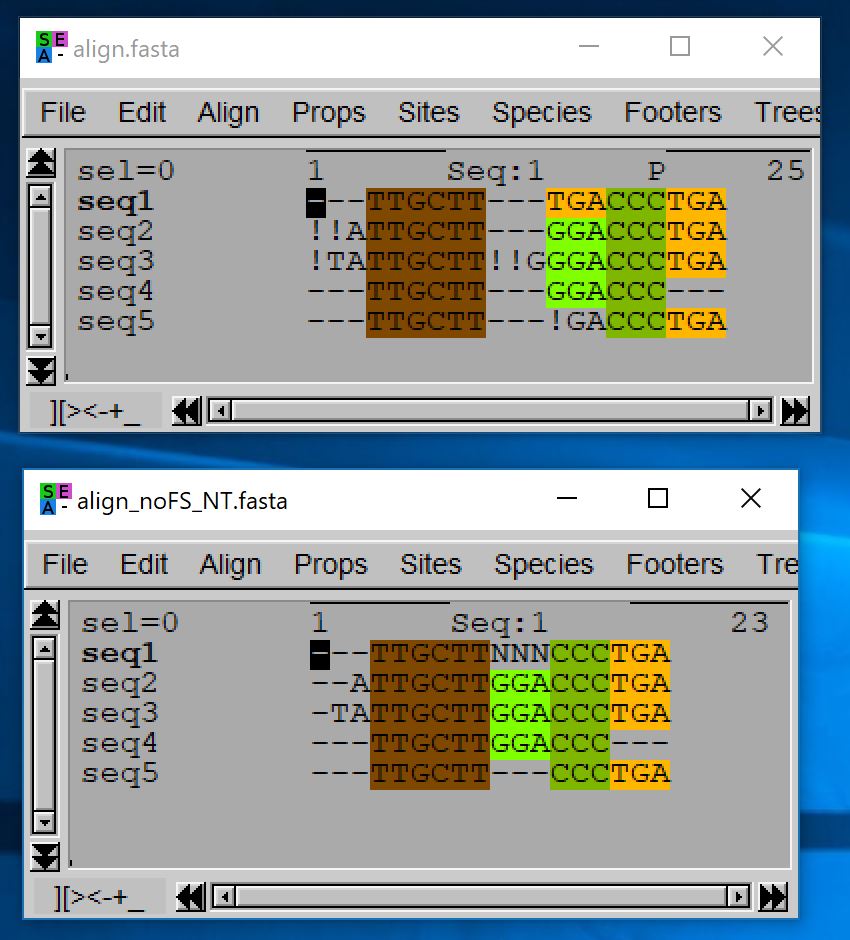
Export guidelines: a reasonable export solution is to preserve final stop codons (as they are expected), to replace other stop codon by « NNN », to replace unexpected internal frameshift codons by « — » (or « NNN ») and to replace remaining frameshift characters (those at sequence extremities) by « -« .
- java -jar macse.jar -prog exportAlignment -align align.fasta -codonForInternalStop NNN -codonForInternalFS — -charForRemainingFS – -out_NT align_noFS_NT.fasta -out_AA align_noFS_AA.fasta
2. Getting some statistics on your (exported) alignment
The following option creates a tabular CSV file containing the number of A, C, G, T, -, and ! characters per site. This could be useful to estimate, for instance, GC or GC3 contents.
- java -jar macse.jar -prog exportAlignment -align align.fasta -out_stat_per_site output_frequencies.csv
You can also obtain a tabular CSV file containing the number of internal frameshifts, stop codons, and deletions for each sequence (the counts are done at the codon/AA level).
- java -jar macse.jar -prog exportAlignment -align align.fasta -out_stat_per_seq output_stats.csv
Those options can be used simultaneously with other export options (e.g. codonForInternalFS). In such a case, the statistics are computed after applying the requested replacements.
3. Having a unique codon for each amino acid
You can use exportAlignment to canonize your alignment. In this case, your alignment will be transformed so that each amino acid will be consistently encoded with the same codon. This could be useful to generate a consensus nucleotide sequence reflecting amino acid frequencies if the consensus solution provided by exportAlignment does not exactly fit your need.
- java -jar macse.jar -prog exportAlignment -align align.fasta -canonize_ON
4. Computing a consensus sequence
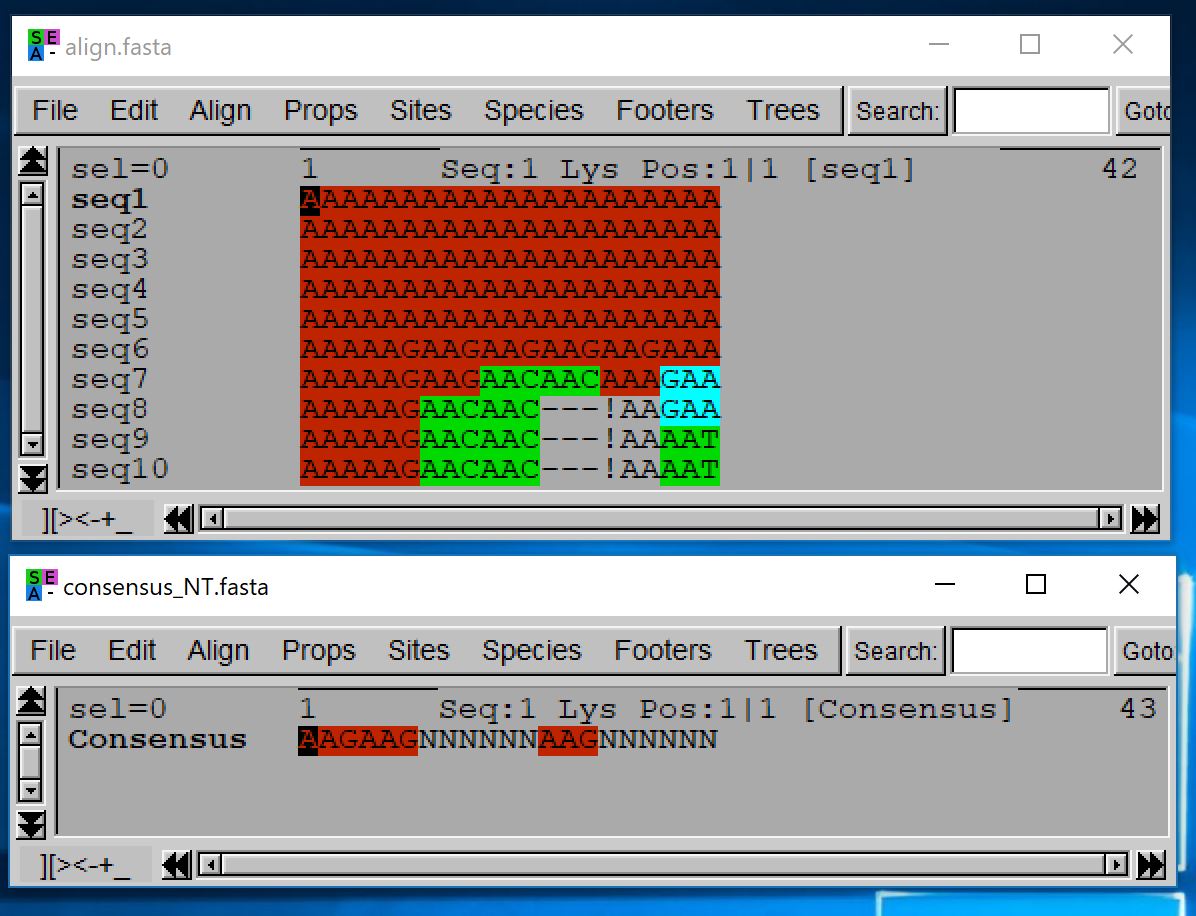
A consensus sequence is a way to summarize an alignment into a single sequence. At the amino acid level, such a sequence can be obtained by keeping for each site the most frequent amino acid. However, if even the most frequent amino acid is not that frequent, you may prefer to have an unknown amino acid ‘X’ at this position. MACSE lets you choose the minimum frequency (percentage) that an amino acid should reach to be kept in the consensus sequence. It also provides a nucleotide consensus sequence such that, if you translate it using the default genetic code, you get the consensus amino acid sequence of your alignment.
- java -jar macse.jar -prog exportAlignment -align align.fasta -cons_threshold 0.71 -out_NT_consensus consensus_NT.fasta -out_AA_consensus consensus_AA.fasta -name_cons_seq Consensus
The name_cons_seq option allows to specify the name of the consensus.
The figure below shows the input alignment provided to the above command and the resulting consensus sequence. Note that with a threshold of 0.71 and 10 sequences, an amino acid has to appear in at least 8 sequences to be present in the consensus sequence. The Aspargine can be encoded by AAA or AAG. The first and second amino acids are thus preserved in the consensus sequence, since in both cases the same amino acid is present in 100% of the sequences (though encoded using two different codons in the second case). The codon used in the consensus nucleotide sequence is the codon used by MACSE to encode Aspargine every time it appears in a consensus sequence (not necessarily the most frequent codon at this position of the input alignment). The fifth amino acid is also preserved as Aspargine is present 6 times out of 7 at this position (gaps are ignored in the frequency computation) and 6/7>0.71.
Finally, for the last three positions of the alignment, the most frequent nucleotides (AAT) each appear in more than 80% of the sequences. However, no amino acid appears in more that 70% of the sequences. This illustrates the difference between a consensus sequence built at the nucleotide level that will return AAT and the consensus at the amino acid level done here by MACSE that returns NNN.
5. Related documentation
You can find other options related to this program from the following links: