MACSE
Adding sequences to a previously computed alignment
Folder: samples/enrichAlignment/
If you have previously computed a protein-coding nucleotide alignment respecting the reading frame, you can use the enrichAlignment subprogram to (conditionally) add new sequences to this alignment. The options are the same as those existing for alignSequences. You can (1) specify a subset of less reliable sequences for which you can define different costs for their frameshifts and stop codons (-seq_lr), (2) choose the name of the output files (out_NT, out_AA), (3) specify the adequate genetic code (gc_def), or (4) select your own elementary alignment costs alignment costs.
1. Basic usage
The basic usage with a single type of sequences is:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta
and if you need to specify that some sequences are less reliable (e.g. newly sequenced, or pseudogenes):
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -seq_lr sequences_lr.fasta
By default enrichAlignment adds sequences to an alignment (referred as the initial alignment) in a sequential mode: each sequence is aligned with the current alignment that contains the sequences of the initial alignment plus those previously added. Some options of enrichAlignment allow you to specify sequences that should be discarded and/or that all the new sequences should be aligned with the unmodified initial alignment (see below).
2. Selecting the sequences to add and those to discard
For each sequence to add, the number of stop codons, frameshifts, codon insertions and codon deletions that will be needed to align this sequence with the current alignment is counted and stored in a tabular file (see below for field details). Those counts may be used to define criteria that the additional sequences should fulfill to be actually kept in the final alignment. For instance, sequences that, once aligned, contain too many stop codons (e.g. > 0), too many deletions (e.g. > 5), too many frameshifts (e.g > 2) or too many internal insertions (e.g >0) could be automatically discarded, using the following options:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -maxSTOP_inSeq 0
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -maxDEL_inSeq 5
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -maxFS_inSeq 2
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -maxINS_inSeq 0
In the latter example, any new sequence with at least one codon insertion will be discarded. Note that this option only counts internal gaps insertions, i.e. those that will required to insert gaps in the middle of the current alignment. If the tested sequence only has longer 3′ or 5′ ends it will be kept.
The total number of codon insertions (internal or not) can also be bounded using:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -maxTotalINS_inSeq 1
All these event upper bound options accept any positive integer as parameters and can be combined to provide a relatively fine tuning of the sequences to be added:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -maxSTOP_inSeq 0 -maxDEL_inSeq 5 -maxFS_inSeq 2 -maxINS_inSeq 0
WARNING
This criteria is not sufficient to ensure that a sequence is actually homologous to those within the input alignment. Verifying that sequences are homologous and can be added to the alignment must be done before using enrichAlignment, for instance using usearch/uclust programs or trimNonHomologousFragments (a subprogram of MACSE developed for this purpose). Indeed, a short sequence of few amino acids will, in most cases, be aligned without the need to introduce frameshifts or stop codons and will not be eliminated. Conversely an homologous sequence may contain frameshifts or stop codons.
3. Fixed alignment and parallelization
Working with a fixed alignment is especially convenient when dealing with (meta)barcoding data since this usually requires to deal with numerous highly similar sequences that are not expected to have indels. When using this option, all sequences to be added are compared with the same initial alignment. The key advantage is that this allows to parallelize the task. For example, if you have 50,000 contigs/sequences that you want to add to your initial alignment, you can split this large dataset in 50 sets of 1,000 sequences each and run the task on 50 computers/CPUs. Moreover, if each of your 50,000 sequences can be correctly aligned with the original alignment without inserting gap events in this original alignment, then the aligned version of your 50,000 sequences (that were computed independently) can be merged to your initial alignment to get a valid global alignment. To ensure this property, we automatically set maxINS_inSeq=0 to identify the subset of the sequences that matches this condition (i.e. no insertion allowed).
– Allowing two different kinds of frameshifts with a fixed alignment
A sequence to add can have two kinds of frameshifts: those where 1 or 2 nucleotides are missing and those where 1 or 2 extra nucleotides are present. The first ones does not impede a sequence to be added to a fixed alignment, whereas the second does. To allow the insertion of sequences having just one frameshift with 1 or 2 nucleotides, those 1 or 2 nucleotides could simply be deleted from the sequences. This is not done by default (as alignment normally do not modify input sequences) but you can allow it using the following options:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -fixed_alignment_ON -new_seq_alterable_ON
– Allowing to trim a few nucleotides at the extremities of the sequences
Sequence extremities are often of lower qualit and this can lead to insertion/deletion near sequences extremities. You can allow MACSE to trim a few nucleotides at the sequence extremities to avoid those insertion/deletion events by specifying the maximal number of nucleotide that can be trimed at each end of the sequences:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -fixed_alignment_ON -max_NT_trimmed 1
– Concatenation of aligned sequences
If you have split the sequences you want to add into smaller subsets, and used enrichAlignment to add them (independently) to your initial alignment, you will need afterwards, to concatenate all your aligned sequences. This will be much easier if each output file only contains the added sequences of the corresponding subset (and not also the initial alignment that would be, by default, in each output file of enrichAlignment), as allowed by the option:
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -fixed_alignment_ON -output_only_added_seq_ON
4. A realistic example related to metabarcoding
Metabarcoding analysis often requires to handle thousands of sequences. Such datasets are not directly tractable with the alignSequence subprogram of MACSE, but they can be handled by sequentially adding your newly obtained sequences to a reference alignment containing sequences of related taxa for your targeted locus (COX1, matK, rbcL, etc…). We successfully used this approach in the Moorea project, M. Leray et al 2013. As we had a initial alignment, used in previous work, we started by using refineAlignment to identify some potential errors in this alignment.
The sequences from the initial alignment are grouped and are given a name starting with OTU whereas added sequences have a name starting with G6YZT. Here the analysis is done on a small subset of the original alignment, using rather strict conditions for insertion and default incremental insertion:
- java -jar macse.jar -prog enrichAlignment -align Moorea_BIOCODE_small_ref.fasta -seq Moorea_BIOCODE_small_ref.fasta -seq_lr noctural_diet_sample.fasta -gc_def 5 -fs_lr 10 -stop_lr 10 -maxFS_inSeq 0 -maxINS_inSeq 0 -maxSTOP_inSeq 1
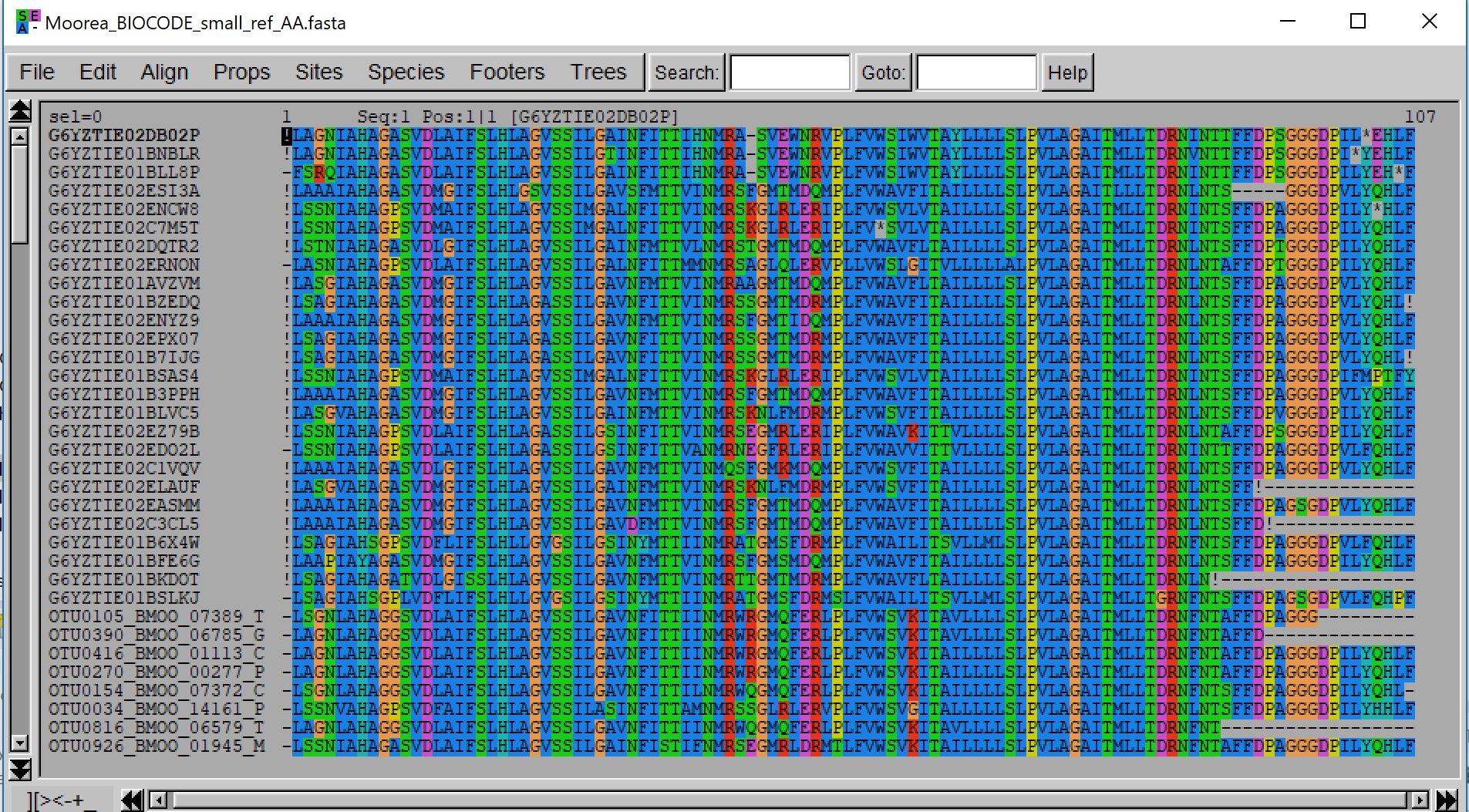
Note that in the above example some of the added sequences induce the insertion of a gap in the original alignment (a gapped codon precedes every OTU sequences). So, if we would have used the fixed alignment option, most of the sequences would have been discarded due to their frameshift on the first codon position. The max_NT_trimmed option is precisely designed to better handle such cases.
On the same dataset, using a fixed alignment (to allow parallelization) and relaxed insertion conditions
- java -jar macse.jar -prog enrichAlignment -align Moorea_BIOCODE_small_ref.fasta -seq Moorea_BIOCODE_small_ref.fasta -seq_lr noctural_diet_sample.fasta -gc_def 5 -fs_lr 10 -stop_lr 10 -maxFS_inSeq 2 -maxINS_inSeq 0 -maxSTOP_inSeq 1 -maxDEL_inSeq 5
leads to the following result (note that a larger number of sequences were added):
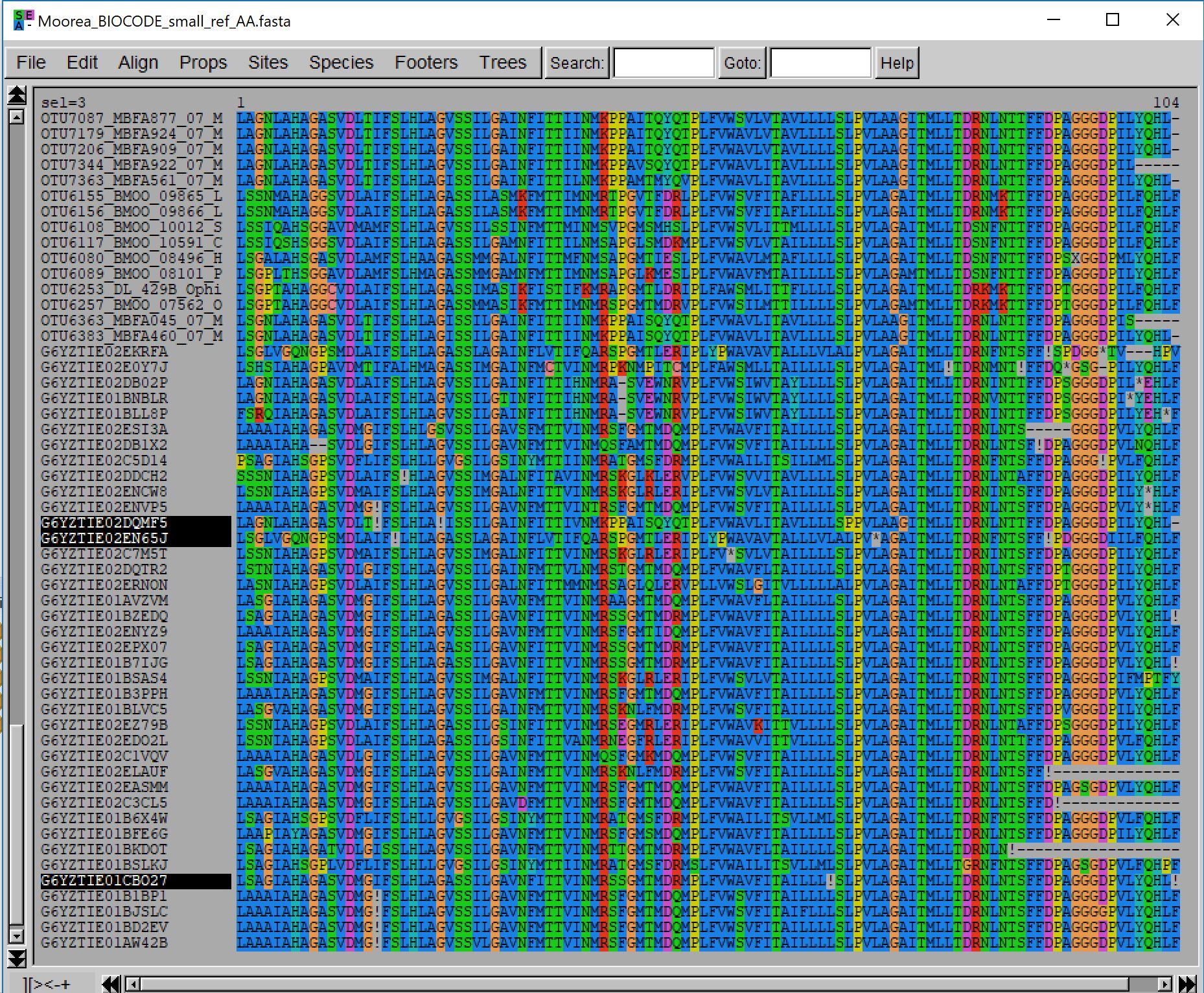
Using these options more sequences were added but some of them contain multiple internal frameshifts and stop codons. Note that even those sequences are correctly aligned by MACSE.
5. Understanding why some sequences are not added and traceability options
The enrichAlignment MACSE subprogram not only produces the two usual FASTA output files containing respectively the nucleotide and amino acid alignments, but also a tabular text file.
- java -jar macse.jar -prog enrichAlignment -align align.fasta -seq sequences.fasta -out_tested_seq_info output_stats.csv
This file contains, for each tested sequence (one per line), the following fields:
- Sequence name
- Whether the sequence was added or not
- Number of frameshifts, trimmed (when sequences are alterable) or not
- Number of trimmed frameshifts
- Number of stop codons
- Number of amino acid deletions (in the sequence)
- Number of amino acid insertions (internal only)
- Number of amino acid insertions (internal or at the alignment extremities)
If you wonder why a sequence was not added to an alignment, you can force its insertion by using relaxed parameters that will allow multiple frameshifts, stops and insertion/deletion events and visualize its alignment.
6. Other options
You can find other options related to this program from the following links :