MACSE
Triming non-homologous fragments before alignment
The TrimNonHomologousFragments subprogram was developed to specifically filter long insertions that often result from annotation errors of introns introduced in CDSs or from alternative splicing. Indeed, positioning long insertions in one or several sequences could drastically slow down the alignment process. Long indels may olaso often prove finally useless since they are removed by alignment filtering tools in subsequent analyses. Finally, non-homologous fragments are often not correctly aligned as indels (see example below) and would hence drastically influence the inferred dN/dS values.
When a compatibility graph of Maximum Exact Match (MEM) is constructed between two genomic sequences, they can be aligned after identification of the longest weighted path Hohl et al. 2002. We extended this approach to handle the translation of nucleotide sequences in the three possible coding frames using a compressed amino acid alphabet. This allowed identifying and suppressing long insertions present in only few sequences before sequence alignment, as such regions, are part of few optimal, or nearly optimal, MEM paths.
Warning This programs mainly aims at removing long non-homologous fragments. Smaller ones will be kept, to avoid removing fragments that are really homologous at this step. If your analyses are sensitive to such non-homologous fragments (e.g. dN/dS estimation), we strongly advice to also use a post filtering of your alignment at the amino acid level (e.g. using HMMCleaner, BMGE or trimAl) and to report this AA masking/filtering at the nucleotide level using reportMaskAA2NT.
1. Simple trimming of non-homologous sequence fragments
- java -jar macse.jar -prog trimNonHomologousFragments -seq ENSG00000125812_GZF1_raw.fasta -out_trim_info output_stats.csv
Although this trimming is done on unaligned nucleotide sequences and returns unaligned nucleotide sequences, we display below the aligned translation of those sequences (using MUSCLE) to illustrate the relevance of this process:
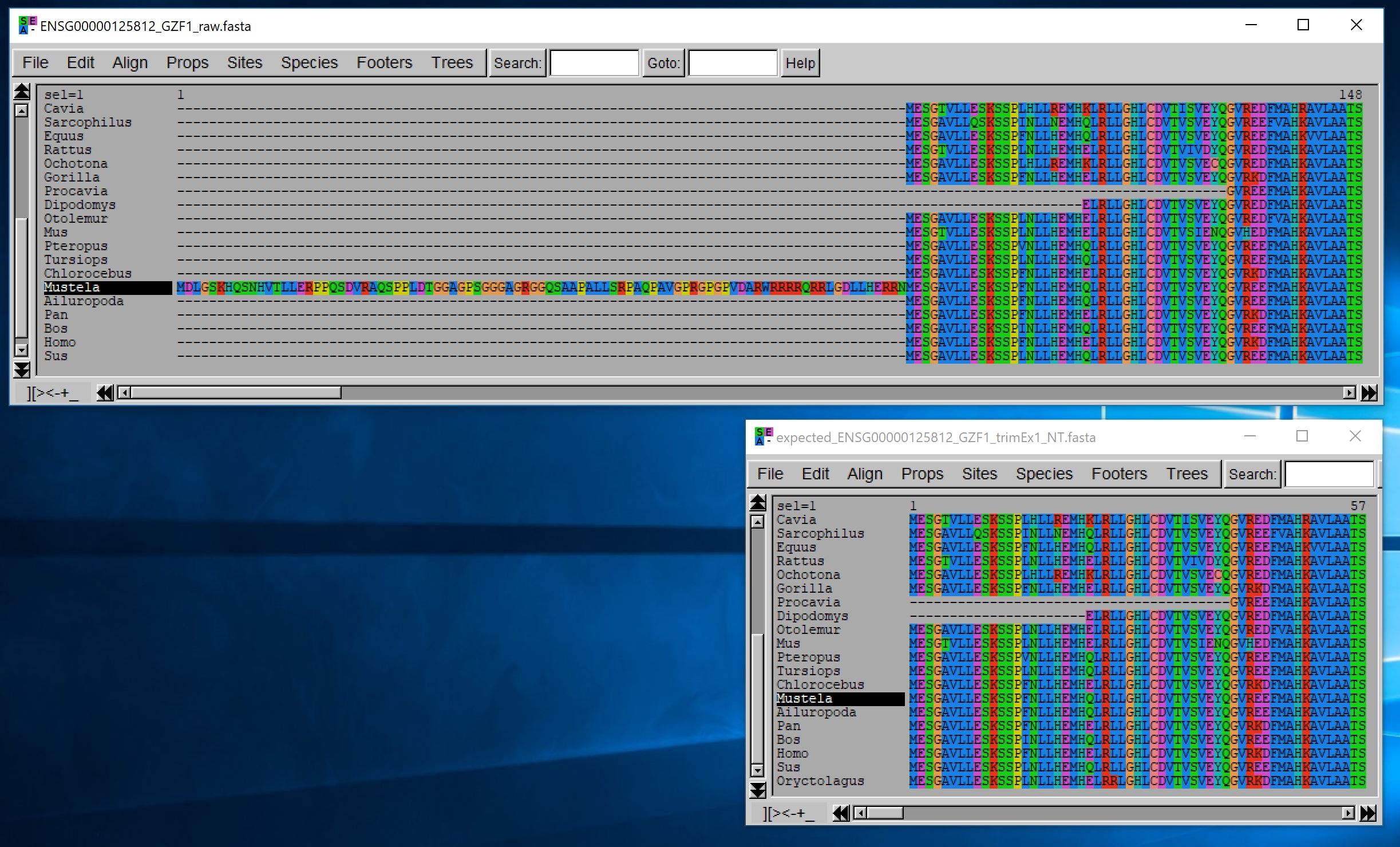
You can control this filtering using different options, but the key ones are the minimal length that a non-homologous fragment should have to be removed when it occurs within a sequence, and when it occurs at a sequence extremity.
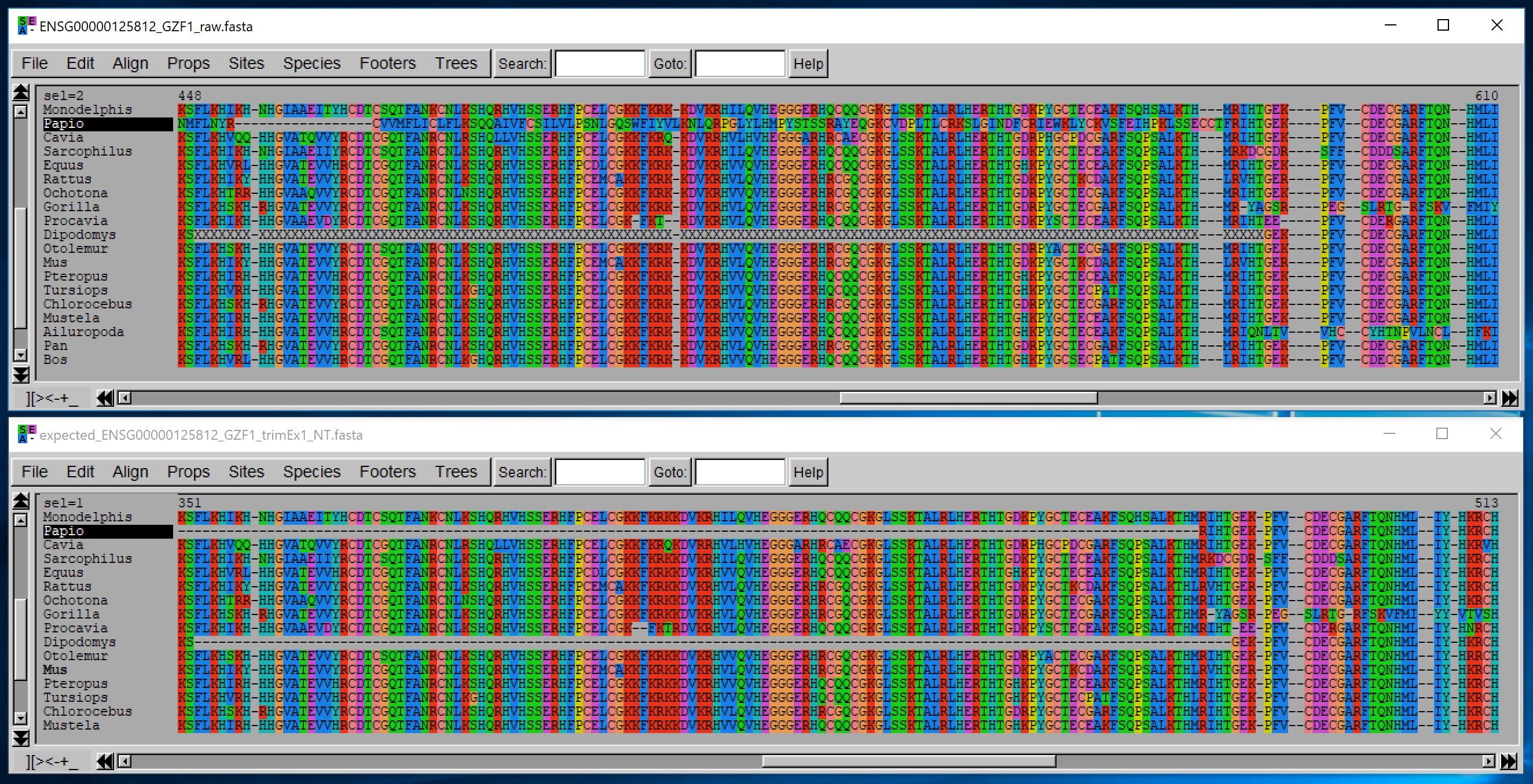
- java -jar macse.jar -prog trimNonHomologousFragments -seq ENSG00000125812_GZF1_raw.fasta -out_trim_info output_stats.csv -min_trim_in 40 -min_trim_ext 20
The result of this trimming is compared with the trimming obtained using default options based on the alignment of their amino acid sequences:
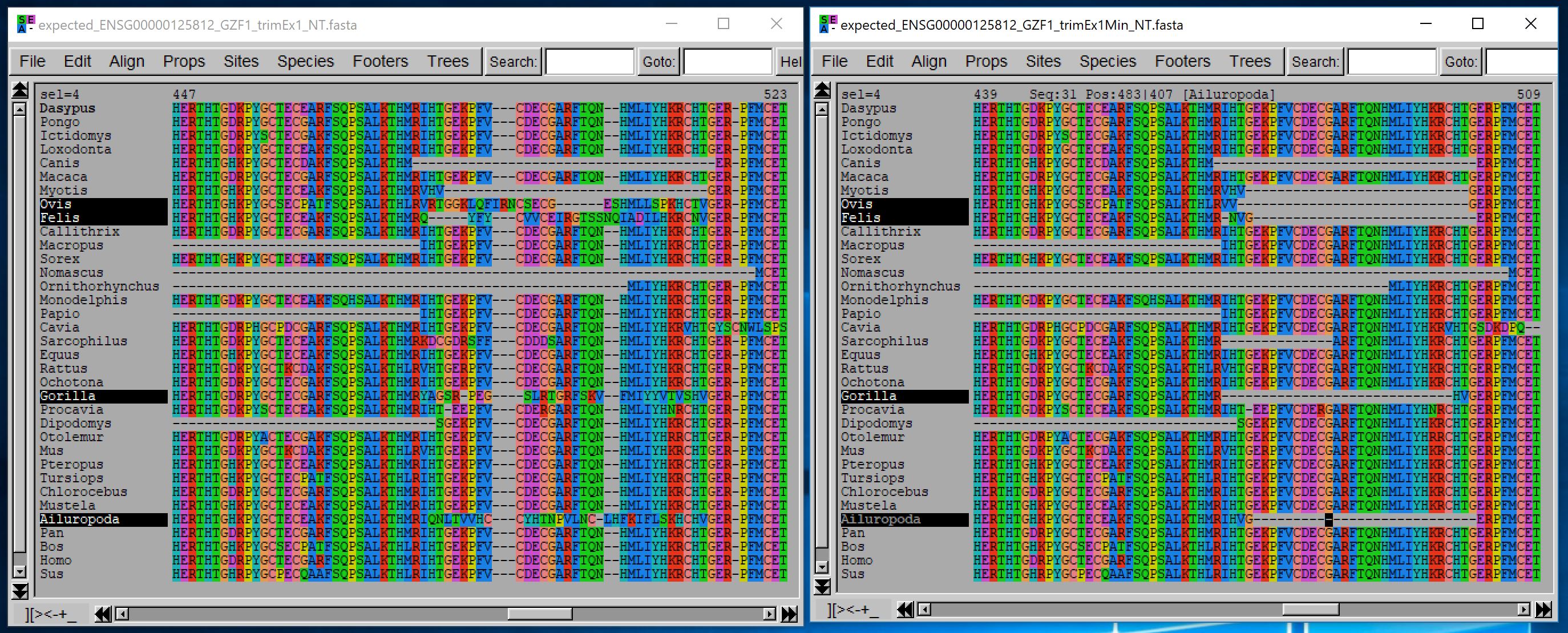
2. Removing sequences that are non-homologous and understanding the output statistic file
If most sites in a sequence are trimmed in this the process, this sequence is likely not homologous to others and it could be better to remove it. You can decide which fraction of the sequence should remain after trimming for a sequence to be kept in the output fasta file:
- java -jar macse.jar -prog trimNonHomologousFragments -seq ENSG00000125812_GZF1_raw.fasta -out_trim_info output_stats.csv -min_homology_to_keep_seq 0.6 -min_trim_in 40 -min_trim_ext 20
A tabular file providing information about the trimming process is provided with one line per sequence and the following columns:
- seqName: the sequence name
- initialSeqLength: the initial sequence length (before trimming)
- nbKeep: the number of nucleotides/characters that remains after trimming
- nbTrim: the number of nucleotides/characters that have been removed by the trimming process (including non informative nucleotides ‘N’)
- nbInformativeTrim: the number of informative nucleotides/characters that have been removed by the trimming process (excluding non informative nucleotides ‘N’)
- percentHomologExcludingExtremities: once extremities have been trimmed which fraction of the remaining part of the sequence has also been trimmed
- percentHomologIncludingExtremities: which fraction of the sequence has also been trimmed
- keptSequences: is the sequence kept and included in the output fasta file
You can specify the name of this output CSV file:
- java -jar macse.jar -prog trimNonHomologousFragments -seq sequences.fasta -out_trim_info output_stats.csv
3. Trimming gappy alignment extremities
To trim only alignment extremities and remove gappy alignment extremities you can use exportAlignment or trimAlignment.
4. Related documentation
You can find other options related to this program from the following links: