MACSE
Report amino acids mask on nucleotides sequences
If your analyses are sensitive to alignment errors (e.g. dN/dS estimation), we strongly advice to use a post filtering of your alignment at the amino acid level (e.g. using HMMCleaner, BMGE or trimAl) and to report this AA masking/filtering at the nucleotide level using.
This subprogram is dedicated to this task. It uses a nucleotide alignment and a filtered (masked) version of its amino acid translation to derived the filtered version of the input nucleotide alignment.
Warning By default some post processings are done to also mask isolated codons (those surrounded only by gaps or masked codons) and sequences with not enough remaining codons can also be completely removed from the alignment.
Here is an example with some of the key options of this program. The input file is a fasta file with the aligned nucleotide sequences and the masked version of the corresponding amino acid alignment. The masking has been done with HMMCleaner, the $ symbol indicate the amino acids masked by HMMCleaner:
- java -jar macse.jar -prog reportMaskAA2NT -align_AA ENSG00000086967_MYBPC2_mask_AA_Hmm10.aln -align ENSG00000086967_MYBPC2_NT_base.aln -min_NT_to_keep_seq 30 -mask_AA $ -min_seq_to_keep_site 4 -min_percent_NT_at_ends 0.3 -dist_isolate_AA 3 -min_homology_to_keep_seq 0.3 -min_internal_homology_to_keep_seq 0.5 -out_stat_file stat.csv
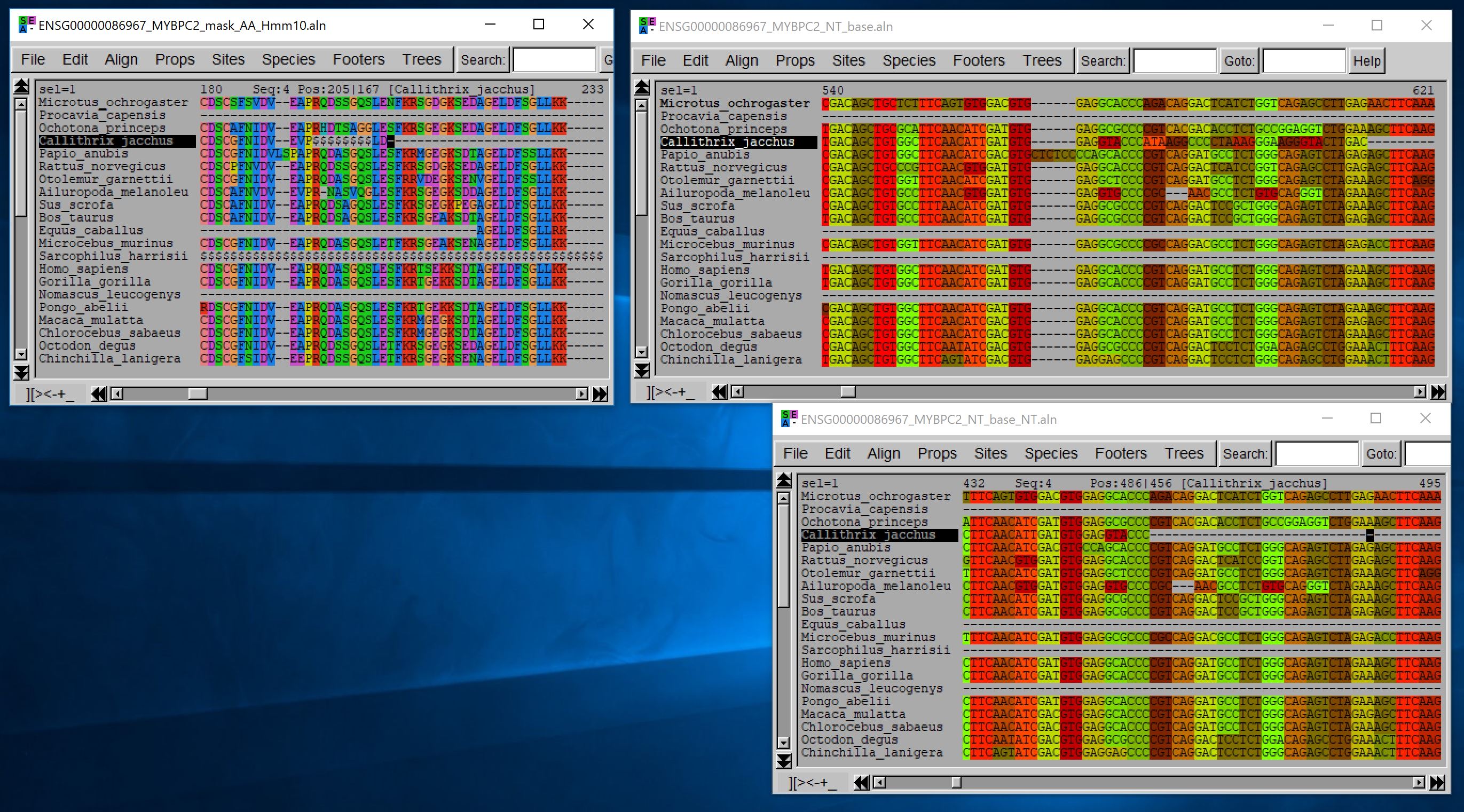
2. Masking traceability
A FASTA file containing the detail of the masking process is also output. In this FASTA unmasked nucleotide are in capital letters while masked ones are in lower case.
3. Related documentation
You can find other options related to this program from the following links:
- allowed nucleotides
- trimAlignment: trims the input alignment by removing gappy sites at the beginning/end of the alignment
- trimNonHomologousFragments: identifies sequence fragments that do not share homology with other sequences and remove those fragments.