MACSE
Alignment trimming
Because protein-coding sequence extremities may contain some UTR fragments, could be less reliable due to errors in the sequencing process, or start at different positions because of different PCR primers being used, alignment extremities are often gappy and therefore not very reliable part of alignments.
Warning This program only uses the number/fraction of gap per position to trim sites. If you want to identify internal sequence fragments that are not homologous or misaligned see trimNonHomologousFragments and reportMaskAA2NT subprograms.
1. Trimming gappy extremities of a nucleotide alignment
This subprogram allows to remove gappy positions from alignment extremities. Starting from one end of the alignment, sites are removed until a site with a large enough number (or percentage) of nucleotides is reached.
- java -jar macse.jar -prog trimAlignment -align align.fasta -min_NT_at_ends 1
- java -jar macse.jar -prog trimAlignment -align AMBN_all_mafft_refined.fasta -min_percent_NT_at_ends 0.8
The figure below displays the impact of the above trimming process on the end of the AMBN sequence alignment (a similar impact is observed at the beginning of the alignment).
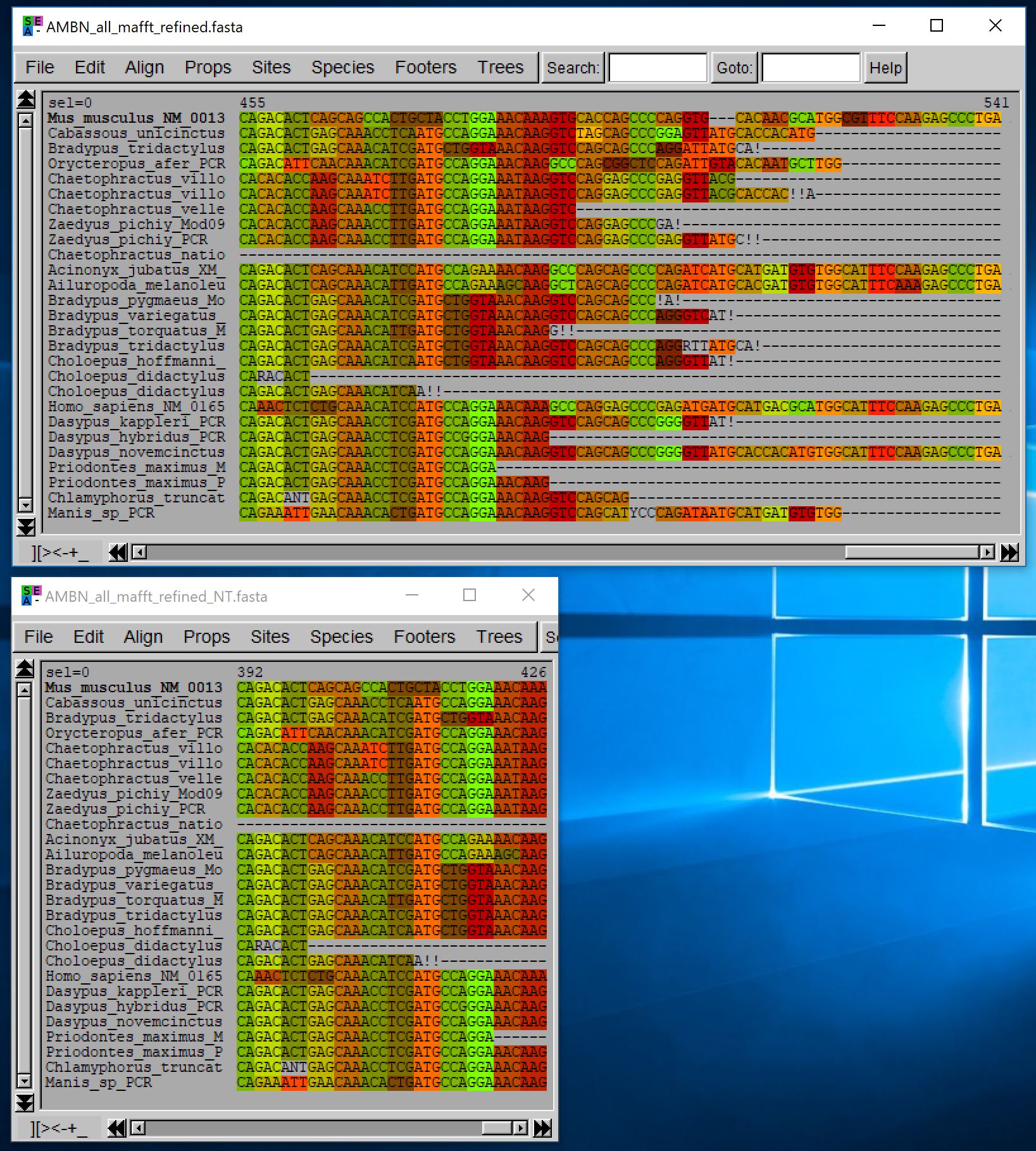
In some cases, an isolated non-gappy site could stop the trimming process. To account for this possibility, you can consider the number/proportion of gaps in a sliding windows instead of considering a single site. The sliding window is centered on a site position and considers the x previous and x following sites. If the window contains enough nucleotides, the central site is the position that will determine one of the two trimming limits (one at the beginning, one at the end of the alignment). We call x the half_window_size.
The default trimming process considers a single site, which is equivalent to using a sliding windows of size 1 resulting from a half_window_size of 0.
- java -jar macse.jar -prog trimAlignment -align align.fasta -min_percent_NT_at_ends 0.25 -half_window_size 3
2. Trimming traceability and output statistic file
A tabular file providing information about the trimming process may be produced if required:
- java -jar macse.jar -prog trimAlignment -align align.fasta -min_percent_NT_at_ends 0.51 -out_trim_info output_stats.csv
Each line of this file provides information regarding the impact of the trimming process on a sequence by providing:
- seqName: the sequence name
- nb_trimed_begin: the number of nucleotides trimmed at the beginning of this sequence
- nb_trimed_end: the number of nucleotides trimmed at the end of this sequence
- trimed_begin: the fragment of the nucleotide sequence trimmed at the sequence beginning
- trimed_end: the fragment of the nucleotide sequence trimmed at the sequence end
3. Related documentation
You can find other options related to this program from the following links:
- allowed nucleotides
- reportMaskAA2NT: uses a NT alignment and a filtered (masked) version of its AA translation to derived the NT alignment.
- splitAlignment: splits an alignment to extract a subset of given sequences and/or sites.
- trimAlignment: trims the input alignment by removing gappy sites at the beginning/end of the alignment