MACSE
Spliting alignment or extracting a sub-alignment (subset of species and/or sites)
Given a large alignment, one can be interested in only a subset of the aligned sequences or in a subset of the sites (i.e. some specific regions/domains of the CDS).
The MACSE subprogram splitAlignment is designed to extract sub-alignments you are really interested in while 1/ removing useless sites, i.e. those only made of gaps after the restriction, and 2/ preserving the codon structure in the resulting sub-alignment since « ! » and « – » characters are handled differently.
WARNING The codon structure is preserved only if, following MACSE convention, the input alignment contains frameshift characters (« ! ») and not classical gap characters (« -« ) to indicate frameshifts in the nucleotide alignment.
Folder: samples/splitAlignment/
1. Extracting an alignment containing a subset of sequences
The subset option allows you to specify which species should be kept in the sub-alignment by taking as argument a text file containing their names. Each line of this text file must contain the name of a sequence, preceded or not by the « > » symbol:
- java -jar macse.jar -prog splitAlignment -align full_seq_set.fasta -subset liste_spe.txt
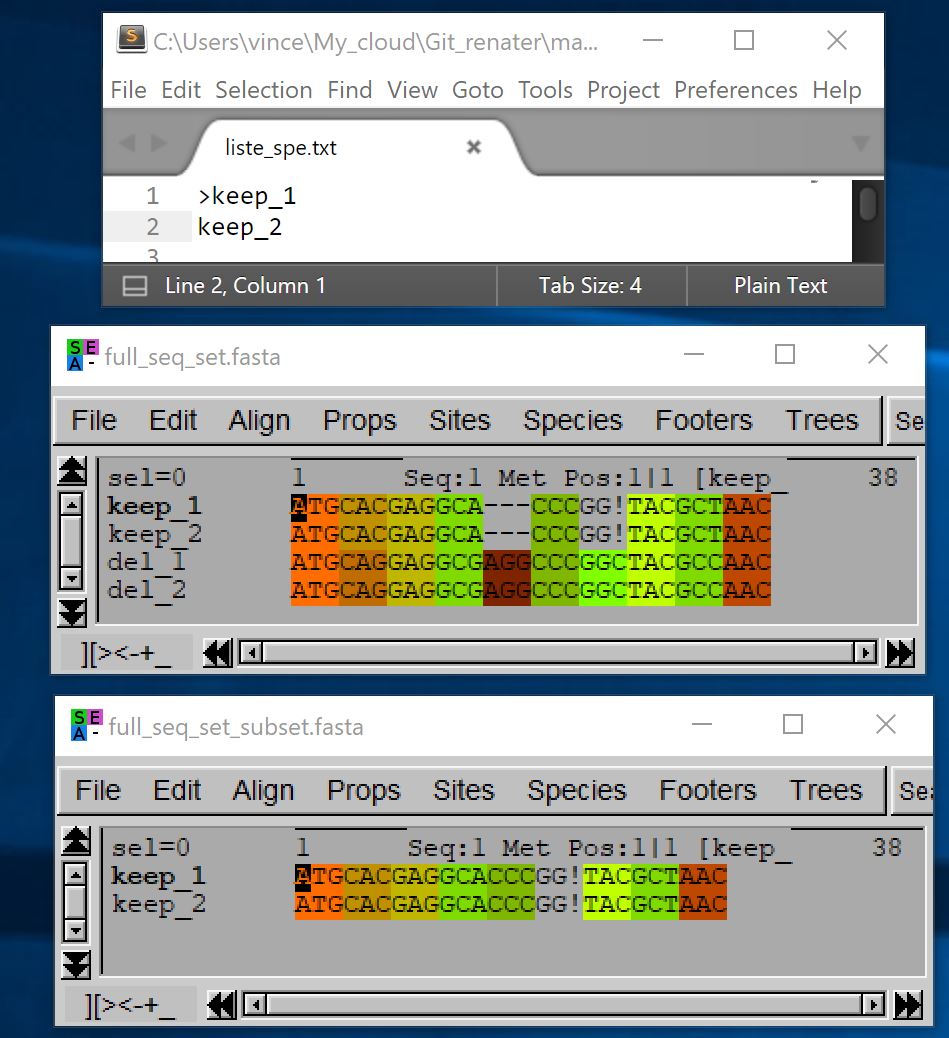
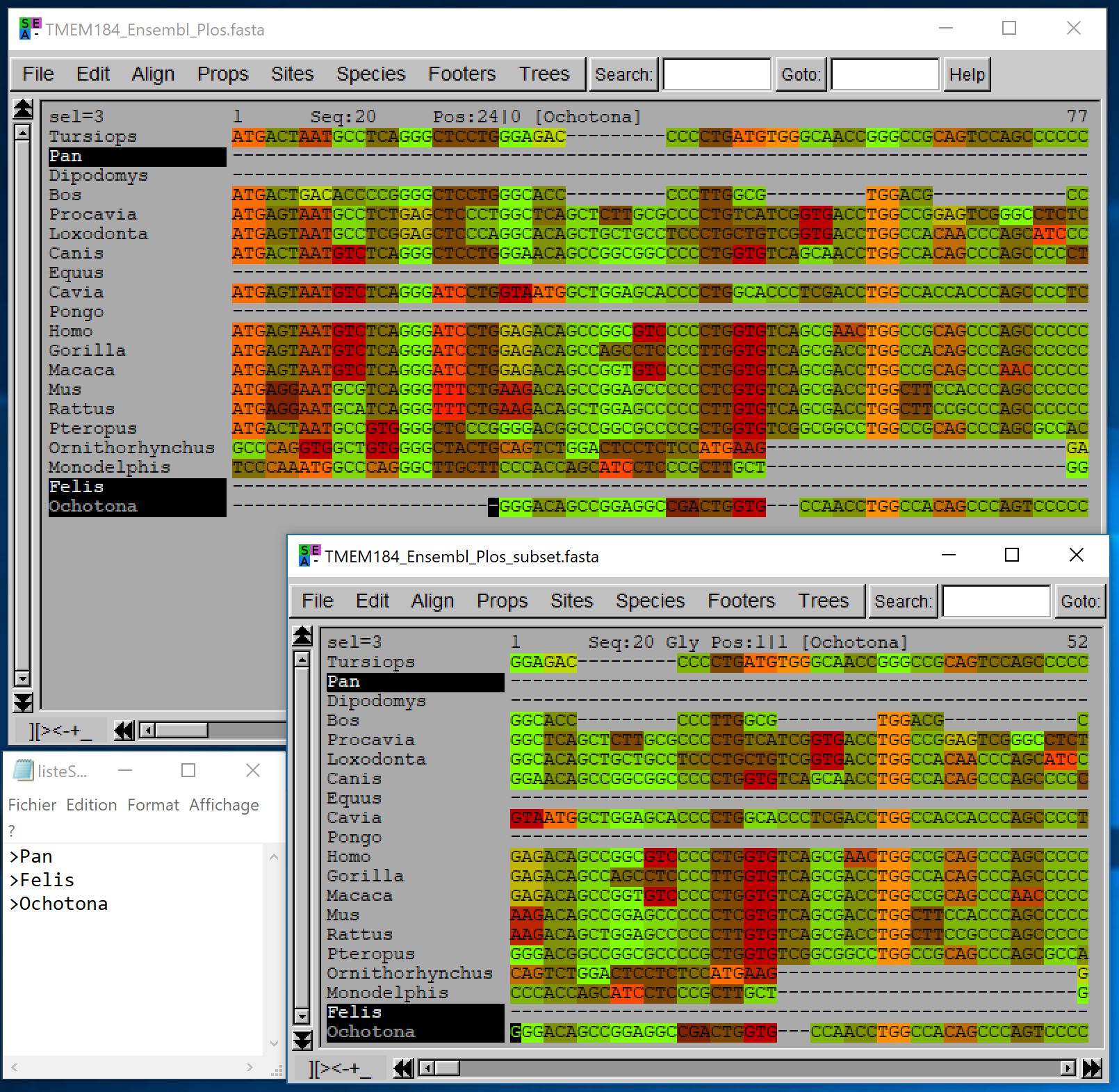
This could be especially useful when you download alignments from public databases such as EnsEMBL or OrthoMaM for which you don’t have a precise control over the species included. For instance if you download an alignment of 1:1 orthologous mammalian sequences from OrthoMaM, you can easily restrict this alignment to Primates by providing the primate species names in a separate file (in OrthoMaM alignments, sequences are named according to the species they come from):
- java -jar macse.jar -prog splitAlignment -align TMEM184_Ensembl_Plos.fasta -subset primates.txt
Note that by default the sub-alignment containing the sequences to kept is saved in a FASTA file whose name end with « _subset.fasta ». The sub-alignment containing all other sequences is also generated and is stored in a FASTA file with a name ending by « _others.fasta ». Of course you can specify different output file names:
- java -jar macse.jar -prog splitAlignment -align align.fasta -subset species.txt -out_subset align_subset.fasta -out_others align_others.fasta
2. Extracting an alignment containing a subset of sites
You can also restrict your alignment to a subset of sites by indicating the region to keep. Here is a basic example :
- java -jar macse.jar -prog splitAlignment -align align.fasta -first_site 4 -last_site 9
If you provide only the first (resp. last) site to keep, the last_site (resp. first_site) will stay fixed at the default value, wich is the end (resp. beginning) of the alignment.
In case you want to preserve several regions of your alignment, you can use a file containing a list of the site intervals to keep:
- java -jar macse.jar -prog splitAlignment -align align.fasta -site_intervals intervals.txt
If it is more convenient to specify the intervals to remove instead of those to keep, you can do so:
- java -jar macse.jar -prog splitAlignment -align align.fasta -site_intervals intervals.txt -reverse_site_selection_ON
- java -jar macse.jar -prog splitAlignment -align align.fasta -first_site 4 -last_site 9 -reverse_site_selection_ON
Instead of providing the interval of sites to keep, using first_site and last_site options, the limits of the interval can be automatically detected on a set of given sequences. In this case, the first_site (resp. last_site) will be the first (resp. last) site for which at least one of the given sequence has a non-gap character at this position. This is convenient when, for instance, you have one or several well annotated sequences mixed with others that may include small UTR fragments (e.g. RNAseq contigs) that you want to remove.
- java -jar macse.jar -prog splitAlignment -align align.fasta -restrict restriction.txt
For more advanced alignment trimming options based on site content see trimAlignment subprogram.
3. Extracting an alignment containing a subset of sequences and sites
You can combine the above options to indicate simultaneously which sequences and sites to keep, e.g.:
- java -jar macse.jar -prog splitAlignment -align TMEM184_Ensembl_Plos.fasta -subset primates.txt -first_site 679
4. Removing sites containing only frameshifts
It is usually preferable to keep sites containing only frameshifts as they preserve the inferred reading frame. However, if you are only interested in having a good nucleotide alignment, you may prefer to consider those sites as containing only gaps and remove them:
- java -jar macse.jar -prog splitAlignment -align align.fasta -keep_FS_OFF
5. Spliting an alignment of amino acids
If you want to split an amino acid alignment you should specify it:
- java -jar macse.jar -prog splitAlignment -align align.fasta -first_site 2 -last_site 3 -amino_alignment_ON